Adverse Events or Unanticipated Problems
The below provides guidance on HHS regulations for the protection of human research subjects at 45 CFR part 46 related to the review and reporting of (a) unanticiaptated problems involving risks to subjects or others; and (b) adverse events. In particular, this guidance clarifies that only a small subset of adverse events occurring in human subjects participating in research are unanticipated problems that must be reported under 45 CFR part 46. The guidance is intended to help ensure that the review and reporting of unanticipated problems and adverse events occur in a timely, meaningful way so that human subjects can be better protected from avoidable harms while reducing unnecessary burden.
*This guidance has been taken directly from the U.S. Department of Health and Human Services
The guidance addresses the following topics:
The HHS regulations at 45 CFR part 46 do not define or use the term adverse event, nor is there a common definition of this term across government and non-government entities. In this guidance document, the term adverse event in general is used very broadly and includes any event meeting the following definition:
Any untoward or unfavorable medical occurrence in a human subject, including any abnormal sign (for example, abnormal physical exam or laboratory finding), symptom, or disease, temporally associated the subject's participation in the research, whether or not considered related to the subject's participation in the research.
Adverse events encompass both physical and psychological harms. They occur most commonly in the context of biomedical research, although on occasion, they can occur in the context of social and behavioral research.
In the context of multicenter clinical trials, adverse events can be characterized as either internal adverse events or external adverse events. From the perspective of one particular institution engaged in a multicenter clinical trial, internal adverse events are those adverse events experienced by the subjects enrolled by the investigator(s) at that institution, whereas external adverse events are those adverse events experienced by subjects enrolled by investigators at other insitutions engaged in the clinical trial. In the context of single-center clinical trial, all adverse events would be considered internal adverse events.
In the case of an internal adverse event at a particular institution, an investigator at that institution typically becomes aware of the event directly from the subject, another collaborating investigator at the same institution, or the subject's healthcare provider. In the case of external adverse events, the investigators at all participating institutions learn of such events via reports that are distributed by the sponsor or coordinating center of the multicenter clinical trials. At many institutions, reports of external adverse events represent the majority of advesre event reports currenlty being submitted by investigators to IRBs.
The phrase "unanticipated problems involving risks to subjects or others" is found but not defined in the HHS regulations at 45 CFR part 46. OHRP considers unanticipated problems, in general, to include any incident, experience, or outcome that meets all of the following criteria:
- unexpected (in terms of nature, severity, or frequency) given (a) the research procedures that are described in the protocol-related documents, such as as the IRB-approved research protocol and informed consent document; and (b) the characteristics of the subject population being studied;
- related or possibly related to participation in the research (in this guidance document, possibly related means there is a reasonable possibility that the incident, experience, or outcome may have been caused by the procedures invovled in the research); and
- suggests that the research places subjects or others at a greater risk of harm (including physical, psychological, economic, or social harm) then was previously known or recognized.
OHRP recognizes that it may be dificult to determine whether a particular incident, experience, or outcome is unexpected and whethere it is related or possibly related to participation in the research. OHRP notes that an incident, experience, or outcome that meets the three criteria above generally will warrant consideration of substantive changes in the research protocol or informed consent process/document or other corrective actions in order the protect the safety, welfare, or rights of subjects or others. Examples of corrective actions or substantive changes that might need to be considered in response to an unanticipated problem include:
- changes to the research protocol initiated by the investigator prior to obtaining IRB approval to eliminate apparent immediate hazards to subjects;
- modification of inclusion or exclusion criteria to mitigate the newly identified risks;
- implementation of additional procedures for monitoring subjects;
- suspension of enrollment of new subjects;
- suspension of research procedures in currently enrolled subjects;
- modification of informed consent documents to include a description of newly recognized risks; and
- provision of additional information about newly recognized risks to previously enrolled subjects.
Only a small subset of adverse events occurring in human subjects participating in research will meet these three criteria for an unanticipated problem.
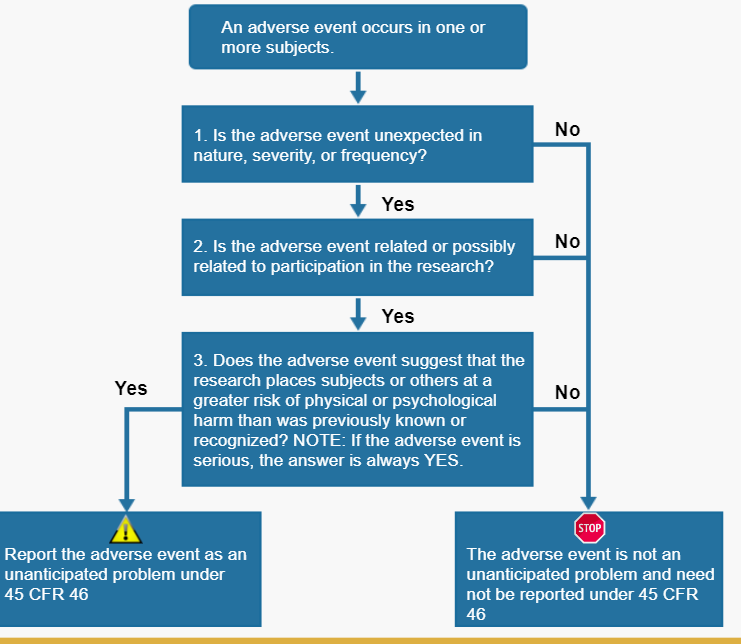
The key question regarding a particular adverse event is whether it meets the three criteria below and therefore represents an unanticipated problem. To determine whether an adverse event is an unanticipated problem, the following questions should be asked:
- Is the adverse event unexpected?
- Is the adverse event related or possibly related to participation in the research?
- Does the adverse event suggest that the research places subjects or others at a greater risk of harm that was previously known or recognized?
If the answer to all three questions is yes, then the adverse event in an unanticipated problem and must be reported to appropriate entities under the HHS regulations at 45 CFR 46.103(a) and 46.103(b)(5).
Below will discuss the assessment of these three questions.
A. Assessing whether an adverse event is unexpected
OHRP defines unexpected adverse event as follows:
Any adverse event occuring in one or more subjects participating in a research protocol, the nature, severity, or frequency of which is not consistent with either:
- the known or foreseeable risk of adverse events associated with the procedures invovled in the research that are described in (a) the protocol-related documents, such as the IRB-approved research protocol, any applicable investigator brochure, and the current IRB-approved informed consent document, and (b) other relevant sources of information, such as product labeling and package inserts; or
- the expected natural progression of any underlying disease, disorder, or condition of the subject(s) experiencing the adverse event and the subject's predisposing risk factor profile for the adverse event.
Examples of unexpected adverse events include the following:
- liver failure due to diffuse hepatic necrosis occurring in a subject without any underlying liver disease would be an unexpected adverse event (by virtue of its unexpected nature) if the protocol-related documents and other relevant sources of information did not identify liver disease as a potential adverse event;
- Hodgkin's disease (HD) ocurring in a subject without predisposing risk factors for HD would be an unexpected adverse event (by virtue of its unexpected nature) if the protocol-related documents and other relavant sources of information only referred to acute myelogenous leukemia as a potential adverse event; and
- liver failure due to diffuse hepatic necrosis occurring in a subject without any underlying liver disease would be an unexpected adverse event (by virtue of its unexpected greater severity) if the protocol-related documents and other relevant sources of information only referred to elevated hepatic enzymes or hepatitis as potential adverse events related to the procedures involved in the research.
In comparison, prolonged severe neutropenia and opportunistic infections occurring in subjects administered an experimental chemotherapy regimen as part of an oncology clinical trial would be examples of expected adverse events if the protocol-related documents described prolonged severe neutropenia and opportunistic infections as common risks for all subjects.
In OHRP’s experience the vast majority of adverse events occurring in the context of research are expected in light of (1) the known toxicities and side effects of the research procedures; (2) the expected natural progression of subjects’ underlying diseases, disorders, and conditions; and (3) subjects’ predisposing risk factor profiles for the adverse events. Thus, most individual adverse events do not meet the first criterion for an unanticipated problem and do not need to be reported under the HHS regulations 45 CFR part 46.103(a) and 46.103(b)(5).
B. Assessing whether an adverse event is related or possibly related to participation in research
Adverse events may be caused by one or more of the following:
- the procedures involved in the research;
- an underlying disease, disorder, or condition of the subject; or
- other circumstances unrelated to either the research or any underlying disease, disorder, or condition of the subject.
In general, adverse events that are determined to be at least partially caused by (1) would be considered related to participation in the research, whereas adverse events determined to be solely caused by (2) or (3) would be considered unrelated to participation in the research.
For example, for subjects with cancer participating in oncology clinical trials testing chemotherapy drugs, neutropenia and anemia are common adverse events related to participation in the research. Likewise, if a subject with cancer and diabetes mellitus participates in an oncology clinical trial testing an investigational chemotherapy agent and experiences a severe hypoglycemia reaction that is determined to be caused by an interaction between the subject’s diabetes medication and the investigational chemotherapy agent, such a hypoglycemic reaction would be another example of an adverse event related to participation in the research.
In contrast, for subjects with cancer enrolled in a non-interventional, observational research registry study designed to collect longitudinal morbidity and mortality outcome data on the subjects, the death of a subject from progression of the cancer would be an adverse event that is related to the subject’s underlying disease and is unrelated to participation in the research. Finally, the death of a subject participating in the same cancer research registry study from being struck by a car while crossing the street would be an adverse event that is unrelated to both participation in the research and the subject’s underlying disease.
Determinations about the relatedness of adverse events to participation in research commonly result in probability statements that fall along a continuum between definitely related to the research and definitely unrelated to participation in the research. OHRP considers possibly related to participation in the research to be an important threshold for determining whether a particular adverse event represents an unanticipated problem. In this guidance document, OHRP defines possibly related as follows:
There is a reasonable possibility that the adverse event may have been caused by the procedures involved in the research (modified from the definition of associated with use of the drug in FDA regulations at 21 CFR 312.32(a)).
OHRP recognizes that it may be difficult to determine whether a particular adverse event is related or possibly related to participation in the research.
Many individual adverse events occurring in the context of research are not related to participation in the research and, therefore, do not meet the second criterion for an unanticipated problem and do not need to be reported under the HHS regulations 45 CFR part 46.103(a) and 46.103(b)(5).
C. Assessing whether an adverse event suggests that the research places subjects or others at a greater risk of harm than was previously known or recognized
The first step in assessing whether an adverse event meets the third criterion for an unanticipated problem is to determine whether the adverse event is serious.
In this guidance document, OHRP defines serious adverse event as any adverse event that:
- results in death;
- is life-threatening (places the subject at immediate risk of death from the event as it occurred);
- results in inpatient hospitalization or prolongation of existing hospitalization;
- results in a persistent or significant disability/incapacity;
- results in a congenital anomaly/birth defect; or
- based upon appropriate medical judgment, may jeopardize the subject’s health and may require medical or surgical intervention to prevent one of the other outcomes listed in this definition (examples of such events include allergic bronchospasm requiring intensive treatment in the emergency room or at home, blood dyscrasias or convulsions that do not result in inpatient hospitalization, or the development of drug dependency or drug abuse).
(Modified from the definition of serious adverse drug experience in FDA regulations at 21 CFR 312.32(a).)
OHRP considers adverse events that are unexpected, related or possibly related to participation in research, and serious to be the most important subset of adverse events representing unanticipated problems because such events always suggest that the research places subjects or others at a greater risk of physical or psychological harm than was previously known or recognized and routinely warrant consideration of substantive changes in the research protocol or informed consent process/document or other corrective actions in order to protect the safety, welfare, or rights of subjects (see examples (1)-(4) in section Appendix D).
Furthermore, OHRP notes that IRBs have authority to suspend or terminate approval of research that, among other things, has been associated with unexpected serious harm to subjects (45 CFR 46.113). In order for IRBs to exercise this important authority in a timely manner, they must be informed promptly of those adverse events that are unexpected, related or possibly related to participation in the research, and serious (45 CFR 46.103(b)(5)).
However, other adverse events that are unexpected and related or possibly related to participation in the research, but not serious, would also be unanticipated problems if they suggest that the research places subjects or others at a greater risk of physical or psychological harm than was previously known or recognized. Again, such events routinely warrant consideration of substantive changes in the research protocol or informed consent process/document or other corrective actions in order to protect the safety, welfare, or rights of subjects or others
For an internal adverse event, a local investigator typically becomes aware of the event directly from the subject, another collaborating local investigator, or the subject’s healthcare provider.
Upon becoming aware of an internal adverse event, the investigator should assess whether the adverse event represents an unanticipated problem following the guidelines described. If the investigator determines that the adverse event represents an unanticipated problem, the investigator must report it promptly to the IRB (45 CFR 46.103(b)(5)).
Regardless of whether the internal adverse event is determined to be an unanticipated problem, the investigator also must ensure that the adverse event is reported to a monitoring entity (e.g., the research sponsor, a coordinating or statistical center, an independent medical monitor, or a DSMB/DMC) if required under the monitoring provisions described in the IRB-approved protocol or by institutional policy.
If the investigator determines that an adverse event is not an unanticipated problem, but the monitoring entity subsequently determines that the adverse event does in fact represent an unanticipated problem (for example, due to an unexpectedly higher frequency of the event), the monitoring entity should report this determination to the investigator, and such reports must be promptly submitted by the investigator to the IRB (45 CFR 46.103(b)(5)).
B. Reporting of external adverse events by investigators to IRBs
Investigators and IRBs at many institutions routinely receive a large volume of reports of external adverse events experienced by subjects enrolled in multicenter clinical trials. These external adverse event reports frequently represent the majority of adverse event reports submitted by investigators to IRBs. OHRP notes that reports of individual external adverse events often lack sufficient information to allow investigators or IRBs at each institution engaged in a multicenter clinical trial to make meaningful judgments about whether the adverse events are unexpected, are related or possibly related to participation in the research, or suggest that the research places subjects or others at a greater risk of physical or psychological harm than was previously known or recognized.
OHRP advises that it is neither useful nor necessary under the HHS regulations at 45 CFR part 46 for reports of individual adverse events occurring in subjects enrolled in multicenter studies to be distributed routinely to investigators or IRBs at all institutions conducting the research. Individual adverse events should only be reported to investigators and IRBs at all institutions when a determination has been made that the events meet the criteria for an unanticipated problem. In general, the investigators and IRBs at all these institutions are not appropriately situated to assess the significance of individual external adverse events. Ideally, adverse events occurring in subjects enrolled in a multicenter study should be submitted for review and analysis to a monitoring entity (e.g., the research sponsor, a coordinating or statistical center, or a DSMB/DMC) in accordance with a monitoring plan described in the IRB-approved protocol.
Only when a particular adverse event or series of adverse events is determined to meet the criteria for an unanticipated problem should a report of the adverse event(s) be submitted to the IRB at each institution under the HHS regulations at 45 CFR part 46. Typically, such reports to the IRBs are submitted by investigators. OHRP recommends that any distributed reports include: (1) a clear explanation of why the adverse event or series of adverse events has been determined to be an unanticipated problem; and (2) a description of any proposed protocol changes or other corrective actions to be taken by the investigators in response to the unanticipated problem.
When an investigator receives a report of an external adverse event, the investigator should review the report and assess whether it identifies the adverse event as being:
- unexpected;
- related or possibly related to participation in the research; and
- serious or otherwise one that suggests that the research places subjects or others at a greater risk of physical or psychological harm than was previously known or recognized.
Only external adverse events that are identified in the report as meeting all three criteria must be reported promptly by the investigator to the IRB as unanticipated problems under HHS regulations at 45 CFR 46.103(b)(5). OHRP expects that individual external adverse events rarely will meet these criteria for an unanticipated problem.
C. Reporting of other unanticipated problems (not related to adverse events) by investigators to IRBs
Upon becoming aware of any other incident, experience, or outcome (not related to an adverse event) that may represent an unanticipated problem, the investigarot should assess whether the incident, experience, or outcome represents an unanticipated problem. If the investigator determines that the incident, experience, or outcome represents an unanticipated problem, the investigator must report it promptly to the IRB (45 CFR 46.103(b)(5)).
D. Content of reports of unanticipated problems submitted to IRBs
OHRP recommends that investigators include the following information when reporting an adverse event, or any other incident, experience, or outcome as an unanticipated problem to the IRB:
- appropriate identifying information for the research protocol, such as the title, investigator’s name, and the IRB project number;
- a detailed description of the adverse event, incident, experience, or outcome;
- an explanation of the basis for determining that the adverse event, incident, experience, or outcome represents an unanticipated problem; and
(4) a description of any changes to the protocol or other corrective actions that have been taken or are proposed in response to the unanticipated problem.
E. Changes to a multicenter research protocol that are proposed by an investigator at one institution in response to an unanticipated problem
For multicenter research protocols, if a local investigator at one institution engaged in the research independently proposes changes to the protocol or informed consent document in response to an unanticipated problem, the investigator should consult with the study sponsor or coordinating center regarding the proposed changes because changes at one site could have significant implications for the entire research study.
F. IRB review and further reporting of unanticipated problems
Once reported to the IRB, further review and reporting of any unanticipated problems must proceed in accordance with the institution’s written procedures for reporting unanticipated problems, as required by HHS regulations at 45 CRF 46.103(b)(5). The HHS regulations at 45 CFR part 46 do not specify requirements for how such unanticipated problems are reviewed by the IRB. Therefore, IRBs are free to implement a wide range of procedures for reviewing unanticipated problems, including review by the IRB chairperson or another IRB member, a subcommittee of the IRB, or the convened IRB, among others. When reviewing a report of an unanticipated problem, the IRB should consider whether the affected research protocol still satisfies the requirements for IRB approval under HHS regulations at 45 CFR 46.111. In particular, the IRB should consider whether risks to subjects are still minimized and reasonable in relation to the anticipated benefits, if any, to the subjects and the importance of the knowledge that may reasonably be expected to result.
When reviewing a particular incident, experience, or outcome reported as an unanticipated problem by the investigator, the IRB may determine that the incident, experience, or outcome does not meet all three criteria for an unanticipated problem. In such cases, further reporting to appropriate institutional officials, the department or agency head (or designee), and OHRP would not be required under HHS regulations at 45 CFR 46.103(a) and 46.103(b)(5).
The IRB has authority, under HHS regulations at 45 CFR 46.109(a), to require, as a condition of continued approval by the IRB, submission of more detailed information by the investigator(s), the sponsor, the study coordinating center, or DSMB/DMC about any adverse event or unanticipated problem occurring in a research protocol.
Any proposed changes to a research study in response to an unanticipated problem must be reviewed and approved by the IRB before being implemented, except when necessary to eliminate apparent immediate hazards to subjects. If the changes are more than minor, the changes must be reviewed and approved by the convened IRB (45 CFR 46.103(b)(4) and 46.110(a)). OHRP recommends that for multicenter research protocols, if the IRB proposes changes to the protocol or informed consent documents/process in addition to those proposed by the study sponsor, coordinating center, or local investigator, the IRB should request in writing that the local investigator discuss the proposed modifications with the study sponsor or coordinating center and submit a response or necessary modifications for review by the IRB.
Institutions must have written procedures for reporting unanticipated problems to appropriate institutional officials (45 CFR 46.103(b)(5)). The regulations do not specify who the appropriate institutional officials are. Institutions may develop written procedures that specify different institutional officials as being appropriate for different types of unanticipated problems. For example, an institution could develop written procedures designating the IRB chairperson and members as the only appropriate institutional officials to whom external adverse events that are unanticipated problems are to be reported, and designating the Vice President for Research as an additional appropriate institutional official to whom internal adverse events that are unanticipated problems are to be reported by the IRB chairperson.
G. Reporting unanticipated problems to OHRP and supporting agency heads (or designees)
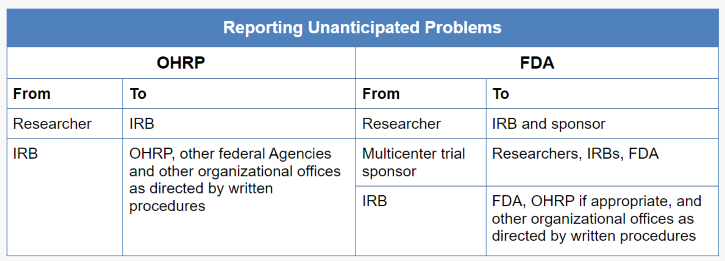
Unanticipated problems occurring in research covered by an OHRP-approved assurance also must be reported by the institution to the supporting HHS agency head (or designee) and OHRP (45 CFR 46.103(a)). Typically, the IRB chairperson or administrator, or another appropriate institutional official identified under the institution’s written IRB procedures, is responsible for reporting unanticipated problems to the supporting HHS agency head (or designee) and OHRP. For further information on reporting to OHRP, see the Guidance on Reporting Incidents to OHRP.
For multicenter research projects, only the institution at which the subject(s) experienced an adverse event determined to be an unanticipated problem (or the institution at which any other type of unanticipated problem occurred) must report the event to the supporting agency head (or designee) and OHRP (45 CFR 46.103(b)(5)). Alternatively, the central monitoring entity may be designated to submit reports of unanticipated problems to the supporting agency head (or designee) and OHRP.
The HHS regulations at 46.103(b)(5) require written procedures for ensuring prompt reporting of unanticipated problems to the IRB, appropriate institutional officials, any supporting department or agency head (or designee), and OHRP. The purpose of prompt reporting is to ensure that appropriate steps are taken in a timely manner to protect other subjects from avoidable harm.
The regulations do not define prompt. The appropriate time frame for satisfying the requirement for prompt reporting will vary depending on the specific nature of the unanticipated problem, the nature of the research associated with the problem, and the entity to which reports are to be submitted. For example, an unanticipated problem that resulted in a subject’s death or was potentially life-threatening generally should be reported to the IRB within a shorter time frame than other unanticipated problems that were not life-threatening. Therefore, OHRP recommends the following guidelines in order to satisfy the requirement for prompt reporting:
- Unanticipated problems that are serious adverse events should be reported to the IRB within 1 week of the investigator becoming aware of the event.
- Any other unanticipated problem should be reported to the IRB within 2 weeks of the investigator becoming aware of the problem.
- All unanticipated problems should be reported to appropriate institutional officials (as required by an institution’s written reporting procedures), the supporting agency head (or designee), and OHRP within one month of the IRB’s receipt of the report of the problem from the investigator.
OHRP notes that, in some cases, the requirements for prompt reporting may be met by submitting a preliminary report to the IRB, appropriate institutional officials, the supporting HHS agency head (or designee), and OHRP, with a follow-up report submitted at a later date when more information is available. Determining the appropriate time frame for reporting a particular unanticipated problem requires careful judgment by persons knowledgeable about human subject protections. The primary consideration in making these judgments is the need to take timely action to prevent avoidable harms to other subjects.
Before research is approved and the first subject enrolled, the investigator(s) and the IRB should give appropriate consideration to the spectrum of adverse events that might occur in subjects. In particular, in order to make the determinations required for approval of research under HHS regulations at 45 CFR 46.111(a)(1), (2), and (6), the IRB needs to receive and review sufficient information regarding the risk profile of the proposed research study, including the type, probability, and expected level of severity of the adverse events that may be caused by the procedures involved in the research. The investigator also should describe how the risks of the research will be minimized.
In addition, depending upon the risks of the research and the likelihood that the research could involve risks to subjects that are unforeseeable, the IRB must ensure, if appropriate, that the research includes adequate provisions for monitoring the data collected to ensure the safety of subjects (45 CFR 46.111(a)(6)). Such provisions typically would include monitoring, among other things, adverse events and unanticipated problems that may occur in subjects enrolled in the research. The HHS regulations at 45 CFR part 46 do not require that the IRB conduct such monitoring, and OHRP believes that, in general, the IRB is not the appropriate entity to monitor research.
OHRP notes that adequate monitoring provisions for research, if deemed appropriate by the IRB, might include one or more of the following elements, among others:
- The type of data or events that are to be captured under the monitoring provisions.
- The entity responsible for monitoring the data collected, including data related to unanticipated problems and adverse events, and their respective roles (e.g., the investigators, the research sponsor, a coordinating or statistical center, an independent medical monitor, a DSMB/DMC, and/or some other entity). (OHRP notes that the IRB has authority to observe or have a third party observe the research (45 CFR 46.109(e).)
- The time frames for reporting adverse events and unanticipated problems to the monitoring entity.
- The frequency of assessments of data or events captured by the monitoring provisions.
- Definition of specific triggers or stopping rules that will dictate when some action is required.
- As appropriate, procedures for communicating to the IRB(s), the study sponsor, the investigator(s), and other appropriate officials the outcome of the reviews by the monitoring entity.
The monitoring provisions should be tailored to the expected risks of the research; the type of subject population being studied; and the nature, size (in terms of projected subject enrollment and the number of institutions enrolling subjects), and complexity of the research protocol.
For example, for a multicenter clinical trial involving a high level of risk to subjects, frequent monitoring by a DSMB/DMC may be appropriate, whereas for research involving no more than minimal risk to subjects, it may be appropriate to not include any monitoring provisions.
For non-exempt research conducted or supported by HHS, the IRB must conduct continuing review of research at intervals appropriate to the degree of risk, but not less than once per year (45 CFR 46.109(e)). At the time of continuing review, the IRB should ensure that the criteria for IRB approval under HHS regulations at 45 CFR 46.111 continue to be satisfied. In particular, the IRB needs to determine whether any new information has emerged – either from the research itself or from other sources – that could alter the IRB’s previous determinations, particularly with respect to risk to subjects. Information regarding any unanticipated problems that have occurred since the previous IRB review in most cases will be pertinent to the IRB’s determinations at the time of continuing review.
It may also be appropriate for the IRB at the time of continuing review to confirm that any provisions under the previously approved protocol for monitoring study data to ensure safety of subjects have been implemented and are working as intended (e.g., the IRB could require that the investigator provide a report from the monitoring entity described in the IRB-approved protocol).
OHRP recommends that, among other things, a summary of any unanticipated problems and available information regarding adverse events and any recent literature that may be relevant to the research be included in continuing review reports submitted to the IRB by investigators. OHRP notes that the amount of detail provided in such a summary will vary depending on the type of research being conducted. In many cases, such a summary could be a simple brief statement that there have been no unanticipated problems and that adverse events have occurred at the expected frequency and level of severity as documented in the research protocol, the informed consent document, and any investigator brochure.
OHRP recognizes that local investigators participating in multicenter clinical trials usually are unable to prepare a meaningful summary of adverse events for their IRBs because study-wide information regarding adverse events is not readily available to them. In such circumstances, when the clinical trial is subject to oversight by a monitoring entity (e.g., the research sponsor, a coordinating or statistical center, or a DSMB/DMC), OHRP recommends that at the time of continuing review local investigators submit to their IRBs a current report from the monitoring entity. OHRP further recommends that such reports include the following:
- a statement indicating what information (e.g., study-wide adverse events, interim findings, and any recent literature that may be relevant to the research) was reviewed by the monitoring entity;
- the date of the review; and
- the monitoring entity’s assessment of the information reviewed.
If an adverse event or unanticipated problem occurs durning your research project, complete the Adverse Event Reporting Form via DocuSign or download the Adverse Event Reporting Form and email to irb@montana.edu
The PI or others involved in research projects will promptly (within 3 calendar days of the event) report to the IRB Chair or Coordinator any substantial changes in activities of the project that was approved by the IRB, any unanticipated problems, unexpected events, or any serious or continuing non-compliance with the study protocol.
The IRB Chair will immediately investigate the situation(s) and may take the following actions:
- If the situation is severe, immediately place the study on "hold" until the MSU IRB can review the information and decide on a course of action.
- Do not place the study on "hold," but refer the situation to the MSU IRB for review.
- If the event posed minimal risk to the subject, deal with the matter administratively.
In cases referred to the MSU IRB, the IRB can:
- Permanently close the study.
- Request that a revised protocol be submitted which contains modified subject eligibility requirements and/or additional safety procedures.
- Decide that the risk to the subject(s) was minimal and let the study proceed, with or without more frequent reviews.
All cases of serious or continuous noncompliance with study protocols will be reviewed by the MSU IRB, which may:
- Permanently close the study.
- Sanction the investigator(s) on all human subject research; sanctions may include suspensions for varying terms or permanent exclusion from participating in human subject research at MSU.
- Institute oversight procedures on the study and/or all studies of the investigator(s).
- Report to the MSU Office of Research Compliance and/or appropriate federal department and agency, if applicable.
How to Determine if the Adverse Event is also an Unanticipated Problem
The criteria necessary to qualify as an unanticipated problem is whether the event is unexpected, related, or possibly related, to participation in the research and places subjects or others at a greater risk of harm. This is not an element in the adverse event definition.